Nessa prática será usado o nível de teoria semi-empíricio PM3 para estudar uma variedade de reações de Diels-Alder. Será necessário executar as seguintes etapas:
A primeira etapa é a otimização
da geometria dos reagentes (etileno,
butadieno, acrilonitrila e isopreno). Primeiro, é
nessário montar as moléculas no
programa MOLDEN,
e salvá-las no formato de Matriz-Z para o progama GAMESS. Usando
o editor PFE edite o arquivo de entrada, input, para o programa GAMESS. Esse
arquivo de entrada deve ter a seguinte estrutura:
$CONTRL RUNTYP=OPTIMIZE COORD=ZMT $END
$STATPT NSTEP=150 HESS=CALC HSSEND=.T. $END
$BASIS GBASIS=PM3 $END
$DATA
(nome do cálculo)
C1
(especificação da geometria)
$END
(obs: apague apenas os textos 'variables' e
'constants', não
excluindo a linha. Além disso, deve haver um espaço em
branco na primeira coluna de cada linha)
Depois é só rodar o cálculo executando o
comando:
C:\DINAMICA\GAMESS.EXE -I EST01.INP -O EST01.OUT
Ele deve durar apenas alguns segundos, gerando um arquivo de
saída, output,
chamado, nesse caso, de EST01.OUT. Quando o cálculo terminar
deve-se
extrair a energia total, e verificar a geometria resultante. Para
visualizar a geometria, abra o programa MOLDEN, clique em "Read", e
selecione o arquivo de saída, output.
Para verificar as geometrias que foram obtidas até a
convergência, clique em "Geom. conv." e/ou no botão
"Movie".
Algumas notas importantes:
Para montar o arquivo de entrada, input, para Etileno:
1.Rode o programa MOLDEN:
C:\DINAMICA\MOLDEN.EXE
2. Clique no botão "Label" e selecione "atom+number"
3. Clique em "ZMAT Editor"
4. Na nova janela "Zmatrix Editor" que se abre, clique em "Add Line"
5. Clique no botão "Double", em seguida clique no átomo
de carbono da janela da tabela periódica que se abre
6. Clique em "Add Line"
7. Antes de marcar o segundo átomo de carbono, clique no
botão "Double", em seguida clique no átomo de carbono, e
clique em C(1) na tela que representa o átomo de carbono
8. Clique em "Add Line"
9. Antes de marcar o átomo de hidrogênio, clique no
botão "Single", em seguida no átomo de hidrogênio,
clique em C(1) e em C(2)
10. Clique em "Add Line"
11. Clique no átomo de hidrogênio da tabela
periódica, em seguida clique em C(1), C(2) e H(3)
12. Continue até completar a estrutura H2C=CH2
13. Salve o arquivo digitando o nome do arquivo, p.ex. EST01.INP, no
campo "File name?"
14. Clique "Write Z-Matrix"
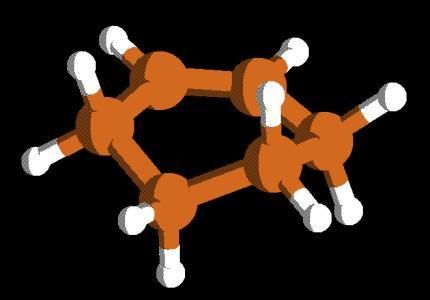
Agora otimize a estrutura do ciclohexeno. Existem várias conformações. A mais estável é similar à figura abaixo:
Essa molécula pode ser montada no programa
MOLDEN conforme os passos abaixo:
1. Rode o programa MOLDEN:
C:\DINAMICA\MOLDEN.EXE
2.Clique em:
ZMAT Editor
Add Line
C <átomo de carbono na Tabela
Periódica>
Substitute Atom by Fragment
-CycloHexane)
3.Clique no último átomo de hidrogênio da matriz Z,
e clique em "Delete Line".
4.Salve o arquivo digitando o nome do arquivo, p.ex. TESTE01.INP, no
campo "File name?"
5.Clique "Write Z-Matrix"
Clique
aqui para voltar ao topo da página.
Encontrar o estado de transição
para uma reação não é uma tarefa simples e
requer alguma experiência com programas de Química
Computacional, como o GAMESS. Eu recomendo que você copie a
geometria do atalho:
Caso você queira tentar encontar a geometria por conta própia, a figura abaixo é uma aproximação de como deve ser o estado de transição:
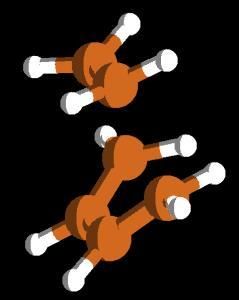
Usando (bastante) o MOLDEN, você
poderá montar uma estrutura similar.
Então, salve o arquivo, e edite o arquivo de entrada para o
GAMESS. Esse arquivo deve ter a seguinte estrutura:
$CONTRL RUNTYP=SADPOINT COORD=ZMT $END
$STATPT NSTEP=150 HESS=CALC HSSEND=.T. $END
$BASIS GBASIS=PM3 $END
$DATA
(nome do cálculo)
C1
(especificação da geometria)
$END
(obs: deve haver um espaço em branco na primeira coluna de cada
linha)
Alguns detalhes desse arquivo de entrada:
Uma vez obtido o estado de transição, você pode comparar essa energia com a dos reagentes e obter uma energia de ativação teórica. Lembrar: as energias estão em unidades atômicas, sendo 1 u.a. = 1 Hartree = 2625,5 kJ/mol.
Clique
aqui para voltar ao topo da página.
O cálculo das freqüências vibracionais já
foi realizado nos cálculos dos reagentes e do estado de
transição, com a inclusão do campo HSSEND=.T. nos
arquivos de entrada. Isso significa o cálculo da matriz de
constantes de força, Hessiana, após a
otimização da geometria. No entanto, para visualizar as
freqüências vibracionais, deve-se fazer um novo
cálculo, seguindo o procedimento:
Feito isso, edite o arquivo TS01-F.INP com a seguinte estrutura:
$CONTRL RUNTYP=HESSIAN COORD=ZMT $END
$BASIS GBASIS=PM3 $END
$DATA
(nome do cálculo)
C1
(especificação da geometria)
$END
(obs: deve haver um espaço em branco na
primeira coluna de cada
linha)
Para visualizar as freqüências
vibracionais e, por exemplo, confirmar se existe uma
freqüência imaginária característica no estado
de transição que representa a passagem de reagentes a
produtos, siga a seqüência:
1. Abra o arquivo de saída TS01-F.OUT no
programa MOLDEN
2. No campo "Frequencies:" clique no botão "Norm. Mode"
3. Vai aparecer uma nova janela com os valores das
freqüências vibracionais harmônicas. Clicando nesses
valores resultará no movimento vibracional característico
para cada
freqüência. Analise a freqüência
imaginária (o primeiro valor)
4. Para verificar, olhe essa animação
do modo vibracional da freqüência imaginária para
o estado de transição da cicloadição
Etileno + Isobuteno
- Etot é a energia
eletrônica total, e é obtida após a
otimização do cálculo (EQUILIBRIUM GEOMETRY
LOCATED), no campo TOTAL ENERGY
- EZPE é a
correção vibracional da energia eletrônica, ou
energia do ponto zero, obtida após a otimização do
cálculo (EQUILIBRIUM GEOMETRY LOCATED), no campo THE HARMONIC
ZERO
POINT ENERGY IS
- Ecorr é a
correção térmica calculada à partir da
termodinâmica estatística, obtida após o campo
THERMOCHEMISTRY AT T= 298.15 K. Abaixo desse campo são
encontrados os resultados
na linha TOTAL para E em kJ/mol ou kcal/mol.
Use as conversões: 1 u.a. = 1 Hartree = 2625,5 kJ/mol =
627,5095 kcal/mol.
Otimize as geometrias dos vários cicloadutos
obtidos a partir dos reagentes substituídos: acrilonitrila e
isopreno. Especificamente, você deve otimizar as geometrias dos
adutos acrilonitrila/butadieno, e as dos dois adutos isoméricos
acrilonitrila/isopreno. A melhor forma de gerar essas geometrias para
esses cálculos é fazer o seguinte:
1. Abra o arquivo de saída, .OUT, do
ciclohexeno menos substituído no MOLDEN
2. Adicione o subsitituinte usando as ferramentas dentro do
botão "ZMAT Editor"
3. Salve as estruturas modificadas
4. Altere esse arquivo usando um editor de texto
5. Rode o cálculo no PCGAMESS.
Note que você deve colocar o grupo ciano em uma posição pseudo-equatorial.
Clique aqui para voltar ao topo da página.
Assim como para os adutos substituídos do ciclohexeno, acima, a melhor forma de iniciar uma otimização de geometria para os estados de transição substituídos e modificar a geometria dos estados de transição convergidos para os estados menos substituídos. Uma vez convergido o estado de transição para uma determinada reação, você pode adicionar o grupo ciano para gerar uma geometria inicial para a reação acrilonitrila + butadieno. Então, quando o estado de transição convergir, adicione um grupo metila na posição meta ou para para gerar a geometria inicial para os dois estados de transição isoméricos para a reação acrilonitrila + isopreno.
Assim como para a reação original, você precisará calcular a matriz de constantes de forças, Hessiana, na geometria inicial.
Nota Importante: Para esses estados de transição, uma nova forma de isomerismo ocorre: o grupo ciano pode ser endo ou exo. Enquando que o produto final no caso considerado aqui não é alterado, a altura da barreira será diferente para os dois modos de aproximação. Aqui você deve considerar apenas os isômeros endo.
Clique aqui para voltar ao topo da página.
Nesse ponto, você já deve ter feito:
Algumas questões para serem trabalhadas:
Clique aqui para voltar ao topo da página.
{kind=link}